
La enfermedad de Fabry es una enfermedad relativamente rara: tiene una incidencia de 1:40 000 a 1:117 000 de nacidos varones. Sin embargo, hay dudas de que esté infradiagnosticada como indican ciertos estudios, debido a la heterogeneidad de fenotipo. Es una enfermedad genética ligada al cromosoma X, que está causada por una mutación en el gen de la enzima alfa-galactosidasa en el cromosoma Xq22. En consecuencia, hay un déficit de dicha enzima y se empiezan a acumular en los tejidos y órganos del cuerpo glicoesfingolípidos, especialmente la globotriaosilceramida (Gb3). La acumulación de estos lípidos tiene lugar en el endotelio vascular, de manera que provocan que los vasos se vayan cerrando hasta producir isquemia y al final la disfunción del órgano.

Estructura de la enzima alfa-galactosidasa
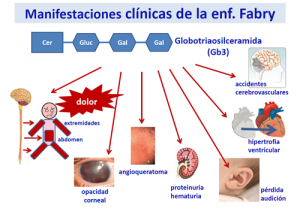
La acumulación de Gb3 en pacientes de Fabry empieza en la etapa prenatal, aunque los síntomas clínicos no se manifiestan hasta más adelante: los más comunes son el dolor agudo en las extremidades, desórdenes gastrointestinales, hipohidrosis. Más adelante, se produce retraso en el aprendizaje y el crecimiento, proteinuria que acaba en fallo renal, cardíaco y enfermedad cerebrovascular. Se sabe que existe una gran variabilidad inter e intrafamiliar entre los síntomas, la edad de comienzo de estos y el curso de la enfermedad. Todo esto dificulta el diagnóstico. Un diagnóstico temprano podría prevenir las complicaciones y la progresión de la enfermedad.
 El tratamiento (aprobado desde 2001 en Europa y 2003 en Estados Unidos) es la terapia de reemplazo enzimático, que consiste en administrar por vía intravenosa la enzima alfa-galactosidasa. Es un tratamiento aprobado en 2001 en Europa y 2003 en Estados Unidos. En Europa, hay disponibles dos preparaciones de enzima a-Gal A (Replagal, Shire Human Genetic Therapies, Boston, MA, USA y Fabrazyme_, Genzyme Corp., Cambridge, MA, USA). A pesar de que ambas proteínas son estructural y funcionalmente muy similares, con la misma secuencia aminoacídica que en la enzima humana nativa, difieren en el patrón de glicosidación de la proteína (que depende de la línea celular originaria). Teóricamente, al solucionar la deficiencia de la enzima se lograría evitar la acumulación de Gb3 y, por tanto, todos los síntomas de la enfermedad de Fabry. Sin embargo, no se ha encontrado una correlación causal directa entre los niveles de Gb3 y las manifestaciones clínicas, al menos hasta la fecha. Así que aunque Replagal y Fabrazyme son eficaces a la hora de disminuir el Gb3, este no es un buen indicador de su eficacia clínica. Se sabe que este tratamiento tiene una eficacia limitada en adultos, por lo que se hipotetiza que si se comienza en niños antes de que desarrollen fibrosis miocárdica, lograría disminuir e incluso prevenir los daños en corazón, riñón y cerebro.
El tratamiento (aprobado desde 2001 en Europa y 2003 en Estados Unidos) es la terapia de reemplazo enzimático, que consiste en administrar por vía intravenosa la enzima alfa-galactosidasa. Es un tratamiento aprobado en 2001 en Europa y 2003 en Estados Unidos. En Europa, hay disponibles dos preparaciones de enzima a-Gal A (Replagal, Shire Human Genetic Therapies, Boston, MA, USA y Fabrazyme_, Genzyme Corp., Cambridge, MA, USA). A pesar de que ambas proteínas son estructural y funcionalmente muy similares, con la misma secuencia aminoacídica que en la enzima humana nativa, difieren en el patrón de glicosidación de la proteína (que depende de la línea celular originaria). Teóricamente, al solucionar la deficiencia de la enzima se lograría evitar la acumulación de Gb3 y, por tanto, todos los síntomas de la enfermedad de Fabry. Sin embargo, no se ha encontrado una correlación causal directa entre los niveles de Gb3 y las manifestaciones clínicas, al menos hasta la fecha. Así que aunque Replagal y Fabrazyme son eficaces a la hora de disminuir el Gb3, este no es un buen indicador de su eficacia clínica. Se sabe que este tratamiento tiene una eficacia limitada en adultos, por lo que se hipotetiza que si se comienza en niños antes de que desarrollen fibrosis miocárdica, lograría disminuir e incluso prevenir los daños en corazón, riñón y cerebro.
El principal inconveniente es el coste elevado de un tratamiento de por vida (por encima de US$200 000 por año en un paciente de 70 kg) junto con la necesidad de administrar repetidamente (cada quince días) grandes cantidades de la enzima alfa-galactosidasa, que además tiene una corta vida media. Algunos pacientes también pueden desarrollar respuestas inmunes contra la terapia.
Actualmente, se están investigando otras opciones terapéuticas como las terapia génica y el papel de las chaperonas. Las chaperonas son pequeñas moléculas que se supone que cruzan la barrera hematoencefálica; lo que no se consigue con la terapia de reemplazo enzimático. Las terapias con chaperonas se administran oralmente, lo que supone una mejora en la calidad de vida del paciente). Aún así, todavía hace falta mucho trabajo antes de que esta investigación se traslade a la clínica.
¿Qué hace un investigador en ciencia de materiales para colaborar en el desarrollo y mejora de fármacos para el tratamiento de enfermedades raras?* Podemos probar a abaratar los costes del medicamento con otros métodos distintos a los actuales que ya están comercializados. Podemos además, intentar mejorar la eficacia de la enzima mediante su encapsulación, para aumentar su actividad y mejorar a nivel celular la internalización de la enzima hacia el lisosoma (recordad que la enfermedad de Fabry es un trastorno lisosomal). Podemos entrar en más detalles más adelante; y si todo va bien, una vez esté publicado puedo hablar más directamente de mi trabajo 😉
*Menciono la ciencia de materiales porque es mi caso, pero también hay muchos especialistas de otras ramas (biólogos, médicos, químicos orgánicos, y un largo etc) implicados en equipos multidisciplinares para tratar de mejorar el tratamiento actual.
Para saber más:
Divulgación buenísima en Curiosidades de la Microbiología
Más técnico en:
Bersano, A., Lanfranconi, S., Valcarenghi, C., Bresolin, N., Micieli, G., & Baron, P. (2012). Neurological features of Fabry disease: clinical, pathophysiological aspects and therapy Acta Neurologica Scandinavica, 126 (2), 77-97 DOI: 10.1111/j.1600-0404.2012.01661.x
Corchero JL, Mendoza R, Lorenzo J, Rodríguez-Sureda V, Domínguez C, Vázquez E, Ferrer-Miralles N, & Villaverde A (2011). Integrated approach to produce a recombinant, His-tagged human α-galactosidase A in mammalian cells. Biotechnology progress, 27 (5), 1206-17 PMID: 21618456
Giannotti, M., Esteban, O., Oliva, M., García-Parajo, M., & Sanz, F. (2011). pH-Responsive Polysaccharide-Based Polyelectrolyte Complexes As Nanocarriers for Lysosomal Delivery of Therapeutic Proteins Biomacromolecules, 12 (7), 2524-2533 DOI: 10.1021/bm2003384
Este artículo ha sido escrito por Dolores Bueno López como colaboración con Raras pero no invisibles. Podéis seguirla en @Ununcuadio o en worlderlenmeyer.blogspot.com.es